

La Mucopolisacaridosis tipo II (MPS II), también denominada Enfermedad de Hunter, es una enfermedad de almacenamiento lisosómico perteneciente al grupo de las mucopolisacaridosis. con afectación multisistémica que conduce a una acumulación masiva de glucosaminoglucanos y una amplia variedad de síntomas que incluyen rasgos faciales gruesos distintivos, baja estatura, afectación cardiorrespiratoria y anomalías esqueléticas.
Epidemiología
La prevalencia de mucopolisacaridosis tipo 2 (MPS2) al nacer varía de 1/72.000 a 1/132.000 de los varones. Es un trastorno recesivo ligado al cromosoma X aunque se han notificado casos muy raros de presentación en mujeres.
Descripción clínica
Se manifiesta como un continuo que varía desde una forma severa (la forma más frecuente) con neurodegeneración hasta una forma atenuada sin afectación neuronal. Los pacientes con MPS2 parecen sanos al nacer, con síntomas iniciales que aparecen entre los 18 meses y los 4 años de edad. La macrocefalia se desarrolla durante la infancia y los bebés inicialmente crecen a tasas normales o superiores a la media. Las manifestaciones iniciales incluyen: infecciones frecuentes del tracto respiratorio (en particular otitis media); hernia umbilical e inguinal; diarrea intratable; hepatoesplenomegalia; y lesiones cutáneas que se asemejan a una cáscara de naranja (en el hombro, la espalda y los muslos). Una facies distintiva con engrosamiento de labios y fosas nasales, así como una lengua agrandada y protuberante que se forma lentamente y puede hacerse evidente entre los 2 y 4 años de edad, o más tarde en casos atenuados. La progresión varía de una forma severa (MPS2, forma severa) con regresión psicomotora temprana a una forma atenuada (MPS2, forma atenuada) que se manifiesta sin compromiso cognitivo.
Etiología
La MPS II está causada por la deficiencia de la enzima iduronato-2-sulfatasa (IDS), lo que provoca la acumulación lisosómica de dos mucopolisacáridos específicos, el sulfato de dermatan (DS) y el sulfato de heparan (HS). El gen causante se ha localizado en Xq28 y se han registrado aproximadamente 320 mutaciones. La MPS II es la única mucopolisacaridosis que se transmite como un rasgo recesivo ligado al cromosoma X.
Aunque teóricamente sólo los varones se ven afectados, se han descrito unos 12 casos de niñas con la enfermedad; en la mayoría de los casos, la inactivación sesgada de uno de los cromosomas X llevó a la expresión preferente del cromosoma X mutado.
Métodos de diagnóstico
El diagnóstico se basa en la presencia de signos clínicos seguidos de la detección de niveles elevados de DS y HS en la orina y se confirma por la demostración de la deficiencia de la enzima en el suero, en los leucocitos o en los fibroblastos, o en muestra de sangre seca. La actividad enzimática de otra sulfatasa también debe evaluarse.
En las mujeres con riesgo de ser portadoras, el análisis de la actividad de la enzima no puede proporcionar una evaluación concluyente de su condición, ya que podría existir inactivación X no aleatoria.
Tests genéticos
Deben someterse a pruebas genéticas cuando la mutación ha sido identificada en el caso índice.
Se estima que las deleciones parciales o las deleciones completas del gen IDS son la causa del 20% de todos los casos. Los pacientes con deleciones completas o grandes reordenamientos del gen IDS tienen en general una presentación clínica severa. Sin embargo, la mayoría de las personas con síndrome de Hunter tienen mutaciones puntuales en el gen IDS. Un pseudogen del gen IDS (IDSP1) se encuentra a poca distancia del gen real. Un evento de recombinación entre el gen IDS y el pseudogen IDSP1 es la causa del aproximadamente el 13% de los pacientes con síndrome de Hunter.
Las pruebas genéticas requieren de la búsqueda de deleciones exónicas o de genes completos, mutaciones puntuales en IDS y su región promotora, y la recombinación con el pseudogen cercano.
La detección de mutaciones puntuales se puede realizar mediante la técnica de secuenciación masiva NGS (exoma clínico dirigido – NGS) mientras que las deleciones exónicas se pueden identificar mediante el uso de la técnica MLPA. En la mayoría de los pacientes cuyo gen sufre un evento de recombinación con el pseudogen IDSP1, esta recombinación produce una inversión, la cual probablemente no sea posible detectar mediante el ensayo de MLPA.
Diagnóstico diferencial
Los diagnósticos diferenciales incluyen mucopolisacaridosis tipo 1, 6, 7; sialidosis tipo 2; mucolipidosis tipo 2 y 3; y deficiencia de sulfatasa múltiple.
Diagnóstico prenatal
El diagnóstico prenatal midiendo la actividad de IDS o mediante análisis de mutación en vellosidades coriónicas o amniocitos sólo se realiza cuando el feto es varón.
Asesoramiento genético
Las mujeres en riesgo de ser portadoras deben someterse a pruebas genéticas ya que MPS2 es recesivo ligado a X. Las mujeres portadoras transmiten el trastorno al 50% de sus hijos; solo se han descrito 12 casos de niñas afectadas debido a una inactivación X sesgada.
Manejo y tratamiento
Además del tratamiento sintomático, que requiere de un enfoque multidisciplinario, la terapia de sustitución enzimática con infusión de la enzima recombinante idursulfasa ha demostrado que alivia los síntomas somáticos aunque sin mejoría de signos neurológicos.
Pronóstico
El pronóstico es muy variable. En la forma severa (60-80% de los casos) la esperanza de vida se reduce notablemente, la muerte generalmente ocurre antes de los 25 años a menudo como resultado de complicaciones cardiorrespiratorias. En la forma atenuada, los pacientes pueden sobrevivir hasta la edad adulta, a veces incluso más allá de los 60 años, y los déficits intelectuales suelen estar ausentes en estos casos.
Prestaciones disponibles en Cibic:
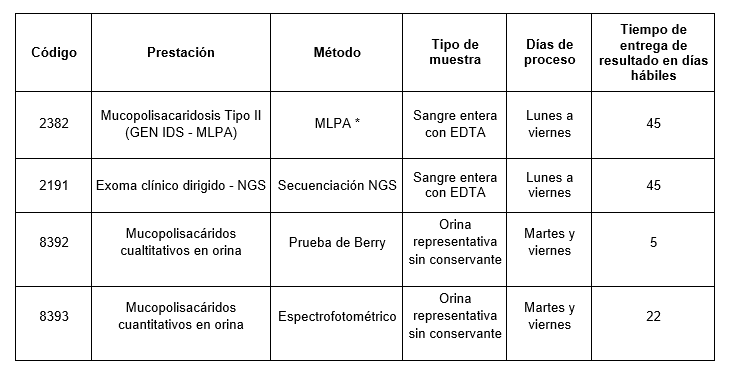
* Detección de deleciones y/o duplicaciones en el gen ID mediante Multiple Ligand-dependent Probe Amplification (MLPA)
Referencias:
– Orphanet. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=580
– Clinical utility gene card for: Mucopolysaccharidosis type II Michael Beck, Frits A Wijburg and Andreas Gal, European Journal of Human Genetics (2012) 20, doi:10.1038/ejhg.2011.143; published online 24 August 2011
– Froissart R, Da Silva IM, Maire I: Mucopolysaccharidosis type II: an update on mutation spectrum. Acta Paediatr Suppl 2007; 96: 71–77.
– Wraith JE, Scarpa M, Beck M et al: Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr 2008; 167: 267–277.
– Martin R, Beck M, Eng C et al: Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008; 121: e377–e386.
– Maurizio Scarpa, MD, PhD. Mucopolysaccharidosis Type II. Gene Reviews. November 6, 2007; Updated: October 4, 2018. https://www.ncbi.nlm.nih.gov/books/NBK1274/.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Alan Gomez.
Tel 0341-4722424. Interno: 243/225
Sección: Análisis Especiales
Lic. Analía Seravalle.
Tel 0341-4722424. Interno: 242.